
MRC Regulatory Support Centre
Version 2, November 2022
1
Retention framework for
research data and records
1 Background and scope
You may be asked to make decisions about the retention of research data and related material
(e.g. laboratory notebooks, biological samples, images, microscope slides, home office licences
and other regulatory or ethical approvals, questionnaires, participant information sheets,
consent forms, etc.).
This framework provides guidance on:
• how to make risk proportionate decisions about the retention or potential destruction of
research data and related material
• the importance of documenting, and keeping a record of, any decisions relating to the
retention (or destruction), of research data and related material; and
• where you can access further guidance to make these decisions with appropriate support.
This framework is designed primarily for UKRI employed staff (e.g. staff in MRC Institutes)
and for those whose clinical research is sponsored by UKRI. It is not designed for researchers
to make decisions about the retention or destruction of research data on their own; but rather
for decisions to be made in partnership, with the involvement of other relevant parties.
Preservation of research data is important for a number of reasons:
• it supports Open Science and promotes reproducibility (important for transparency);
• it provides an audit trail or an archive of the research run in your Institute (this may include
permanent preservation of research which is historically significant or important);
• it enables future research opportunities through data or sample sharing.
However, the retention of research data (including any sample storage / biobanking) and long-
term archiving, does not come without financial cost. It’s therefore important to weigh up the
risks of retaining research data versus the risks of its destruction before taking well-informed,
risk proportionate decisions about what to keep. Efforts can then focus on managing retention
for research which poses ‘high risk’ and taking a light touch approach for all other research.
Examples of research posing ‘high risk’ might include:
• Research studies where there is a greater likelihood that scientists will be asked to provide
data and/or samples, to support further research or to enable reproduction of findings as
these are high profile / high impact or in some way contentious; or
• Research which has generated Intellectual Property (IP), or where there is potential IP; or
• Research studies where there is a greater likelihood that participants might suffer harm as
a result of the research.

MRC Regulatory Support Centre
Version 2, November 2022
2
1.1 MRC Policy
MRC’s Retention Policy for research data and related material is outlined in Good Research
Practice: Principles and Guidelines. Good Research Practice (GRP) is intended to complement
statutory or regulatory requirements (e.g. Clinical Trials Regulations, data protection law,
1
etc.).
The retention periods in GRP apply to research data and related material in MRC Institutes
where MRC / UKRI is your employer or the sponsor of your research. If you are based in MRC
Head Office or in an MRC institute there is additional guidance on managing business
information (i.e. non-research records) in the UKRI Information Management Policy and UKRI
Retention Schedule. You can also seek guidance from the UKRI Knowledge and Information
Management Team.
For MRC funded research, GRP is advisory although it can be used to inform local policy.
Where MRC is purely your funder, local organisational policy and/or your sponsor(s) policies on
retention will apply (e.g. if you do clinical research in a university, then your university and/or
sponsor(s) guidance on retention should be followed).
For MRC university units and centres (where MRC used to be the employer) GRP applies to
research data and related materials generated when MRC was your employer. (Any retention
commitments may be outlined in the Unit closure or Unit transfer agreement). For research data
and related material generated after transition, local university and/or sponsors’ policies apply.
MRC also provide information on Open Research and Data Management and Sharing.
Minimum retention periods for research data and related material where UKRI is your
sponsor and/or employer
For basic research – at least 10 years after the study has been completed.
For population health and clinical studies – at least 20 years after study completion.
Studies which propose retention periods beyond the minimum limits stated above, must include
valid justification. However, it may be appropriate to consider longer retention periods in some
cases; for example, where there is:
• a greater likelihood that scientists will be asked to share data with others for further
research or for reproducibility reasons (i.e. because their research findings are high
profile/high impact or in some way contentious);
• Intellectual Property or the potential for IP to arise (e.g. laboratory notebooks and/or
relevant records from clinical studies could be retained indefinitely);
• a greater likelihood that participants might suffer harm as a result of the research. For
example, a clinical drug trial involving pregnancy (i.e. the clinical trial actively recruited
pregnant mothers or a participant / their partner became pregnant during the trial), then a
retention period of 25 years might be more sensible to cover any potential claim for
1
In terms of primary data protection law, researchers can keep data indefinitely for research (subject to
appropriate ‘safeguards’). If research data are no longer useful, or unlikely to be used in further research, then
you should consider their destruction.

MRC Regulatory Support Centre
Version 2, November 2022
3
damages that may arise from either the participant or their child). For those who lack
capacity to consent, a claim could arise at any time. Therefore, it would seem sensible to
review what is being kept at 20-25 years to determine whether to keep records longer.
• a direct impact on national policy making and/or the results are of historical importance
(e.g. the MRC have permanently preserved proposals relating to the Human Genome
Mapping Project in The National Archives).
1.2 Active management
There should be a clear and transparent system in place to manage research data and
samples.
• There should be an identified person, or persons, responsible for managing all research
data and/or samples (e.g. the Principal Investigator or Trial Steering Committee, or a
librarian, Information / Data Manager, Designated Individual or equivalent).
• There should be a register, database or inventory to manage research samples and/or
data. For example the UKRI Information Asset Register tracks relevant information on key
UKRI datasets such as storage location, time of data collection, type of data and any
decisions with respect to destruction.
• It should be easy for those authorised to access data and/or samples, to be able find and
access what they need (i.e. via meta-data or data documentation for electronic format or
an inventory or database for samples or other physical records). This is particularly
important for sample and data sharing.
1.3 Organisational policies
In order for organisations to carry out their responsibilities, under data protection law as well as
research governance responsibilities
2
, all will have their own policies on data / records retention.
(We discussed MRC’s policy at 1.1).
1.4 Expectations of other organisations
Similarly, funders, journals and others may have their own requirements. You will need to
understand and meet the policies of all of the relevant organisation(s) involved in your research
(e.g. those providing regulatory oversight, your collaborators, funders or journals) and consider
how these expectations will impact research data and/or sample retention. (In some cases
these expectations may be outlined in an agreement).
For example, any organisation providing regulatory oversight may either audit and/or inspect
your premises (e.g. Health & Safety Executive, Home Office, Human Tissue Authority,
Medicines and Healthcare products Regulatory Agency, etc.). You should be aware of the
research data and/or samples that they are likely to want to see and ensure that you can
provide access to these.
2
These are outlined in the UK policy framework for health and social care research. In addition the Medicines for
Human Use (Clinical Trials) Regulations outline requirements for Clinical Trials of Investigational Medicinal
Products.

MRC Regulatory Support Centre
Version 2, November 2022
4
If your research involves external collaboration with the NHS, they will have their own
expectations and may check compliance. You should be aware of these expectations, be
confident that you are in a position to deliver them and consider any long-term requirements
(i.e. how long do they expect you to retain any research data and/or samples and who should
pay for storage?).
2 Deciding what, how and where to keep
2.1 What to keep?
During the lifecycle of any research project a whole range of data and/or samples will be
generated, recorded, collected or used. What to keep will very much depend upon your
research and what you need in order to enable an understanding of what was done when, how
and why. This process should be led by someone who is familiar with the research (e.g. the
Principal Investigator or Trial Steering Committee) with decisions about retention being made in
line with organisational policy and sponsor(s) expectations (where appropriate).
For some basic research the information recorded in laboratory notebooks may provide
sufficient detail to enable an understanding of what was done when, how and why. Whilst for
some clinical studies a whole raft of data / records will be required in order to enable that
same understanding.
Decisions about what to keep need to be made on a case-by-case basis with respect to which
data and/or samples are required in order to meet the aims of data preservation (e.g. to support
open science and reproducibility of findings, to provide an audit trail and to enable data
sharing). Where appropriate any decisions must be in line with the expectations of participants;
i.e. if a participant were to withdraw consent from your study, then you would retain their data
and/or samples in line with what was agreed with respect to their withdrawal (e.g. the participant
may have agreed that you can retain what you already hold, but that you won’t accrue any
further data and/or samples).
A risk proportionate approach should be taken to decisions about retention and any long-term
archiving. There should be some form of risk assessment for all studies to determine those
which are ‘high risk’ and require additional management. For example, you may decide to
manage ‘high risk’ studies by retaining data and/or samples for longer in their original format.
That’s not to say that all research data from ‘high risk’ studies (or indeed any study) should be
kept for longer and/or in their original form as the scenarios below demonstrate:
What to keep – scenario 1
For some basic research any decision-making about what to keep may be influenced by the
outcome of the research. A record of what was done when, how and why will always be kept in
laboratory notebooks. If an experiment didn’t work (rather than produced negative results) then
the original output may not be as valuable to preserve and may not necessarily be kept
(although a record of this decision should be clearly documented, signed off and retained for
future reference so that you can track the fate of the original output). You may also consider
retention of original outputs in the short term for protocol optimisation reasons.

MRC Regulatory Support Centre
Version 2, November 2022
5
Some original outputs take up a lot of physical space (e.g. plates and cell cultures). Normally
plates are captured via a plate reader and the images printed and/or saved electronically.
Keeping a cell culture may also be less important if the initial biological starting material and/or
any resulting cell line is preserved. As such cell cultures and plates would usually only be kept if
deemed useful and/or unlikely to deteriorate.
What to keep – scenario 2
Some outputs can deteriorate over time. Gels and immuno blots are normally photographed and
then discarded as they are too fragile to maintain. Stained microscope slides may be retained
after analysis but may not be worth keeping in the long-term as the staining can fade over time
(and slides can take up a lot of physical space).
What to keep – scenario 3
It’s worth considering whether keeping a physical paper record is necessary in the longer term.
If you were to recruit healthy volunteers to a volunteer database, you might ask them to
complete a paper form to capture some personal details (i.e. name, address, contact telephone
number) as well as the types of research in which they might like to take part. All of these
details could then be entered into a database and the original form stored for reference.
Is it worth keeping these original paper forms in the longer term? The information that you need
is now stored in the database. Therefore, as long as there is some form of quality assurance
process to ensure that the data in the database is accurate and valid, then the original paper
forms can be destroyed after an agreed period of time. Your quality assurance process should
be clearly documented, signed off, and retained for future reference (this should include details
about the destruction of the original paper forms). It’s important to document the decision to
destroy these forms and retain a copy of this decision for future reference. You can then
accurately demonstrate what has been destroyed and track the fate of all your research data
and/or samples.
It’s also worth considering the risk of keeping the original paper forms in the longer term and
balancing this against the risk of destruction. The forms contain ‘personal data’ and are
therefore subject to data protection law and confidentiality requirements. Storing the original
paper forms in the longer term, particularly if this will mean storing them with an external
archiving organisation, presents a potential risk to your volunteers’ privacy as well as a cost
burden.
2.2 What format - Physical versus electronic?
Scenario 3 can be applied more generally as any research data may be collected in physical
form (i.e. biological samples, microscope slides, on paper, etc.) and/or in electronic form. Where
both a physical and electronic copy of the same data exists, decisions can be made with
respect to whether to keep both formats in the longer term or not. These decisions should be
made by someone who is familiar with the research data (e.g. the Principal Investigator or Trial
Steering Committee) in line with organisational policy and sponsor expectations (where
appropriate).

MRC Regulatory Support Centre
Version 2, November 2022
6
Good Research Practice: Principles and Guidelines allows for the conversion of original paper
records into electronic format (e.g. by inputting data into software or by scanning). In principle,
both of these options can be a good solution to reducing long-term paper archiving costs.
However, neither comes without cost, both are time consuming and there is the potential for
error as explored in the following scenario.
Scanning scenario: An MRC Unit wanted to stop retaining the original paper copies of a
screening form and consent form for people undergoing an MRI scan, and switch to electronic
scanned copies. In line with Good Research Practice, as long as there is a system in place
which serves to check that the screening forms and consent forms have been scanned to an
appropriate standard, are held electronically and that this process has been signed off and
documented, then it would demonstrate that the data were: "… validated using quality
assurance procedures" and therefore destruction of the paper copies could be justified. (The
Unit also recorded the decision to destroy the paper forms, and retained this for future
reference, so they could track the fate of these forms).
It's about balancing risk:
• how likely is it that you will need to provide these forms to someone in the future (for
whatever reason – it could be for an audit, a complaint, etc.)?
It is more likely that you will be asked to provide these forms for research considered ‘high
risk’ (i.e. for research, which is high profile, high impact or in some way contentious; or
where there is a greater likelihood that participants might suffer harm).
• if you had to provide these forms would this be easier to do in paper or electronic format?
• what are the risks of retaining paper records versus electronic format? Paper can take up
more physical space than its electronic counterpart. Whilst the potential for things to be
destroyed (e.g. fire, flood) or go missing / be accidently deleted are probably fairly similar
in each case, with electronic format there is the option to make a back-up copy.
However, it’s important to note that storing electronic records is not without cost (e.g. in
terms of scanning time, server space, etc.). Electronic records also need active
management to ensure they remain accessible over time (i.e. maintaining audit trails /
keeping track of back-ups, ensuring security of back-ups, and regularly checking that files
can continue to be accessed and read as software and hardware technology advances,
etc.).
• what are the risks of transferring paper records to electronic format – again, this is where a
process of checking or validation is absolutely vital (it's no use archiving scanned records
if they haven't scanned as intended and you've got incomplete copies).
There is also the possibility for scanned records to be amended (either deliberately or
accidently). Therefore, access to these should be controlled by an appropriate person.

MRC Regulatory Support Centre
Version 2, November 2022
7
2.3 Where to keep?
Initially research data and/or samples will be collected and retained under the supervision of the
Principal Investigator or Trial Steering Committee (i.e. in the Principal Investigator’s employing
organisation, Clinical Trials Unit(s) and/or at any other site(s) also involved in the research).
At some point a decision may be taken to curate the data and/or samples centrally; if so, your
organisation should have a policy or process on when they should be curated centrally and
where.
Storage of biological samples: Existing collections of samples often have significant value for
research. To ensure this potential is maximised, samples can be transferred to a central tissue
resource or ‘biobank’, to maintain quality and optimise sample use. In England, Wales and
Northern Ireland research ‘biobanks’ are subject to the Human Tissue Authority’s licensing
scheme, overseen by a Designated Individual (or equivalent for unlicensed premises). The
Designated Individual is a key contact for sample management.
Samples are often only as valuable as any accompanying data; the following paragraph is
relevant where any accompanying data is identifiable.
Archiving identifiable information: In multi-site studies, participants’ identifiable information
will usually remain at the site at which they were recruited and/or treated. Although, some
identifiers may be shared with the coordinating centre (or biobank) to ensure future linkage or
for participant follow-up. Whatever the arrangements are for a specific study, participants should
be informed (and agree) to how their identifiable information will be used for research.
Completed consent forms contain identifiable information and as such are likely to be kept at
each local site. Sites may incur archiving costs as a consequence of storing such
documentation. These costs should be identified early and arrangements made as to how they
will be covered.
When archiving identifiable information away from the controlled environment in which they
were originally collected and collated, the Principal Investigator or the Trial Steering Committee
should consider any additional risk. With guidance from the sponsor and relevant Data
Protection Officer(s), Principal Investigator(s) or the Trial Steering Committee should ensure
that only the identifiable information that needs to be kept is kept (i.e. a risk-based decision) and
that data security can be maintained.
All data: Wherever research data are kept, their security must be ensured and they must be
maintained / stored in such a way that they can be easily identified and located to support future
activities (including access for audit purposes, data sharing, etc.). If you are considering storing
research data with an external service provider, you must ensure that they can do this.
If you would like further guidance on approved providers of external archiving, please contact
the UKRI Knowledge and Information Management Team.

MRC Regulatory Support Centre
Version 2, November 2022
8
2.4 Reviewing what you keep
Long-term storage should not be the end of the story, your organisation should have a policy /
process for reviewing what is kept with a view to either further retention or destruction of
research data or samples as appropriate. Research data or samples should only be kept for as
long as they are useful (e.g. to support further research, to defend any scientific challenge for
research which is high profile / high impact or in some way contentious, to investigate any
instances of participant harm, etc.). If research data or samples are no longer useful, you should
consider their destruction or disposal. (The Retention decision-making flowchart in Annex 1
provides further guidance).
Reviewing what you keep scenario
The process of review could be initiated by the impending departure of a member of staff from
the organisation (e.g. a PhD student moving on, a programme leader retiring, etc.). Principal
Investigators (PIs) have a key role to play in any decisions about the retention or destruction of
research data and/or samples collected, generated or used within their programme of research.
It is therefore particularly important that PIs are involved in any retention or destruction
decisions before they (or one of their students) moves on to another research organisation, or
the PI retires.
Where a Principal Investigator (PI) has a large collection of biological samples and data, then
it’s important for them to help determine:
• Whether anyone wants these samples and/or data?
o Perhaps the PI accessed the samples and/or data from a biobank or a data provider
and a legal agreement is in place stating the fate of the samples and/or data; or
o The PI or student is keen to take the samples and/or data with them to their new
research organisation; or
o Someone else within the current research team, another collaborator or a biobank
wants to take responsibility (or custodianship) of the data and/or samples;
• Whether the Principal Investigator is confident about the utility of the samples and/or data?
Have the samples been stored appropriately, is there any evidence to prove sample
utility? Similarly, is the data ready for sharing? Does it have appropriate high-quality meta
data? (Any recipients of the samples and/or data are likely to want to be assured of utility);
• Whether there are any restrictions which would limit the use of the samples and/or data?
Are any legal agreements in place which state what can and cannot be done with the
samples and/or data? Do the samples have specific storage requirements (e.g. do they
need to be kept in a -80 freezer?). For both the samples and data, does the consent in
place support the planned secondary use, do you have evidence of consent? (Again, any
potential recipients of the samples and/or data, are likely to want assurance in terms of
consent. Work with local contacts to determine what assurance is most appropriate in your
circumstances).
If the secondary use or retention of the data and/or samples will involve transferring them to
another organisation, then the transfer arrangements should detail any conditions of access and
eventual destruction (these arrangements could be outlined in a legal agreement). Where

MRC Regulatory Support Centre
Version 2, November 2022
9
relevant the Data Protection Officer(s) and/or the Designated Individual (or equivalent) in both
your organisation and the recipient organisation should be notified. You should also ensure that
the decision to transfer the samples and/or data is clearly documented, signed off and retained
for future reference (so that there is an enduring record to track what has happened to the
samples and/or data).
All decisions (i.e. either further retention / transfer, or the destruction of samples and/or data)
should be documented, signed off (by the Unit Director, Head of Department, Designated
Individual (or equivalent), Data Protection Officer(s) or sponsor representative) and kept for
future reference. This will ensure that there is a clear record of the fate of all research data
and/or samples within your organisation.
Good Research Practice: Principles and Guidelines recommends that research data relating to
studies which directly inform national policymaking or are of other historical importance, are
considered for permanent preservation. In some cases the potential impact on policy may be a
clear aim of the study, whilst in others this significance may only come to light later. It may
therefore be necessary to consider the impact of the study at several stages during its lifecycle,
particularly for studies which have long-term goals and run for many years. Once decisions
have been made about what to keep and in what format, what is held should be reviewed on an
ongoing basis. The table below suggests some timelines:
Suggested
retention periods
Potential decisions?
Less than 5 years
Retain physical data in its original form. Unless doing so would
render it unusable, e.g. would microscope slides fade and degrade
in this time?
Between 5-20
years
Make decisions with respect to what will be retained and in what
format. For example you may wish to consider scanning paper
records, or if you have any remaining biosamples, are these still
useful?
For electronic records, it would be timely to consider active
management of software and/or hardware. The impact of software
and/or hardware upgrades becomes more significant over time
and these begin to impact on the ability to access files. Therefore,
systematically checking files and upgrading any software and/or
hardware as necessary, will ensure continued access to your
research data.
More than 20
years
Consider destruction unless the research data are extremely
valuable
e.g. has informed national policy making / are of historical
importance or are from a “risky” participant group (e.g. ‘high risk’
study involving children, adults without capacity or contentious
research outcome).

MRC Regulatory Support Centre
Version 2, November 2022
10
3 Data curation and further use
It’s important to consider how data will be stored, maintained and accessed to ensure it remains
useable and retrievable for secondary or future research (particularly important for electronic
data, i.e. to ensure it can be accessed and continues to be accessible as technology advances).
The Concordat on Open Research Data promotes openly available research data for use by
others wherever possible.
3.1 Where will data be kept?
• Will you (the researcher) keep some/all of the data?
• Will it go to a repository?
• If your research will generate clinical information, will the resulting clinical data form part of
the patient’s medical record?
3.2 Who will curate the data?
• Will you (the researcher) maintain the data and any associated meta-data in a useable
format?
• Will agreements be put in place with another organisation to manage the data on your
behalf?
• How will access for secondary or future research or for reproducibility reasons be
facilitated?
3.3 How will access requests be managed?
• Will access be actively promoted, is the data and/or meta-data discoverable by other
researchers?
• Are data publicly available or available through a managed/controlled access process? For
datasets containing individual participant data MRC supports managed access.
• How are access requests reviewed, which criteria are used to make decisions?
• Will you be able to provide an extraction service by those with knowledge of the data and
storage systems? Will they help new users understand the data, will it be a collaborative
relationship? Long term, how will you retain this expert knowledge of the data?
• In what format will data be sent to the other party? Is secure data transfer required?
• How will you oversee usage, e.g. a data sharing agreement with recipients?
For further information on sharing research data please see MRC data management and
sharing; and for sharing individual participant data, please see MRC Methodology Hubs
guidance Good practice principles for sharing individual participant data from publicly funded
clinical trials.
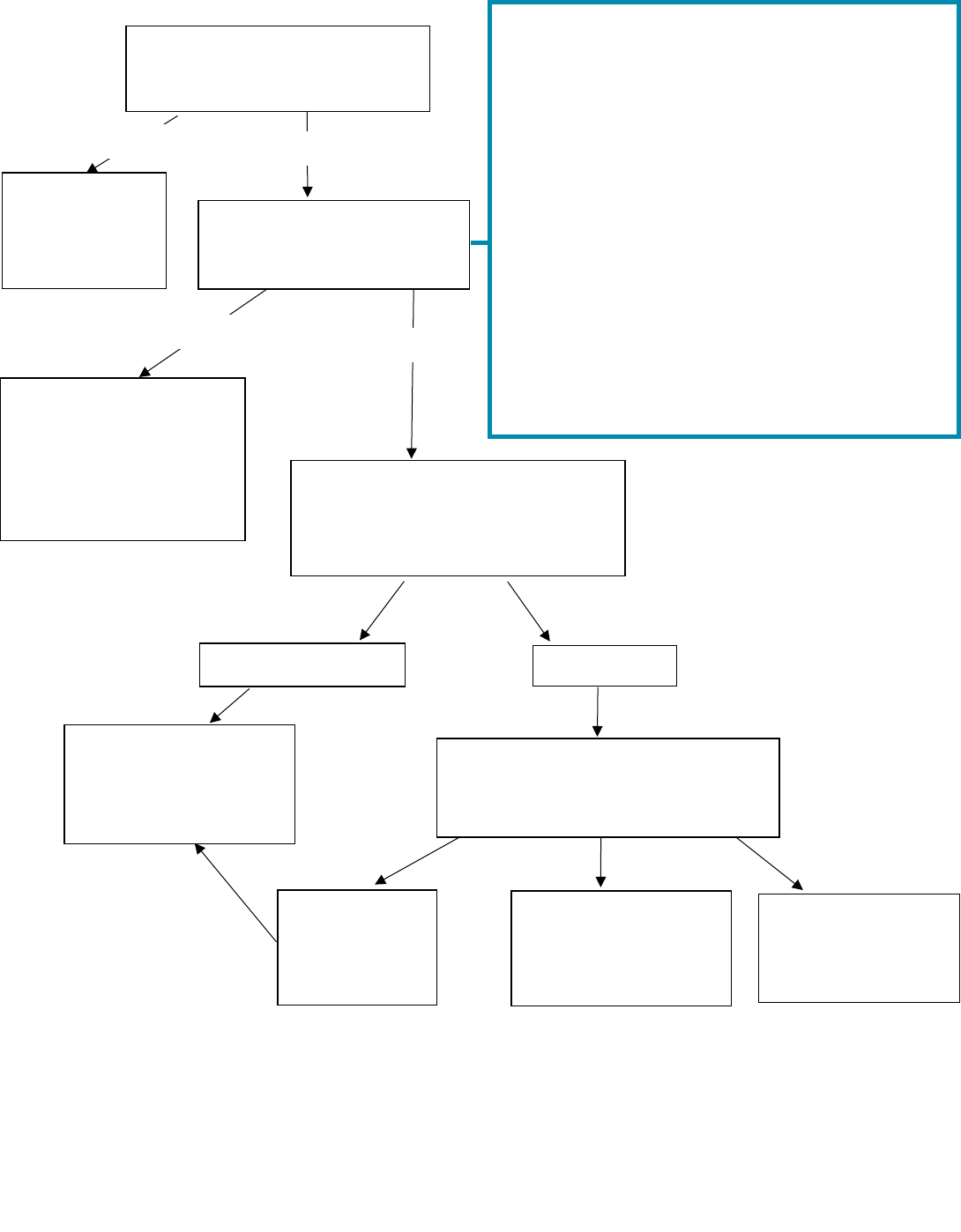
Annex 1: Retention decision making flowchart for research data
and/or samples
MRC Regulatory Support Centre
Version 2, November 2022
11
Comply with
the terms of
the
agreement.
Retain data and/or
samples in line with
your organisation’s
policy
2
or arrange
transfer to the new
custodian
3
.
Your organisation.
Third party.
Consider destruction
or disposal in line
with your
organisation’s policy.
Destroy or dispose
of on behalf of third
party (retain record
of permission).
Return to third
party (retain
record of
decision).
NO
YES
YES
1.
You should consider the effort required to prepare your data and/or samples for sharing, before following
the ‘YES’ route. For example, is your data already collated? Does it have appropriate meta-data?
2.
All organisations should have a retention policy. MRC’s policy is outlined at 1.1.
3.
You should not arrange the transfer of data and/or samples on your own, involve the appropriate local
contacts (e.g. your sponsor representative, Data Protection Officer, Designated Individual as appropriate).
Is an agreement in place
which states the fate of the
data and/or samples?
If no
response or
contact is not
practicable.
NO
Are the data and/or
samples still needed?
(Please see box on right).
Who sponsored the research?
(If there was no sponsor, who
was the Principal Investigator’s
employer?)
Contact third party and obtain
decision over fate of data and/or
samples where practicable.
Things to consider in assessing whether data
and/or samples are still needed
If any of the following are likely, follow ‘YES’ route:
a) You will need to access the data and/or
samples to support further research (i.e. your
own research, or research done by others).
b) A new custodian is found for the data and/ or
samples (e.g. a collaborator, biobank, etc.)
1
c) Your research has/will lead to a high-profile
publication which could be challenged.
d) Your research includes Intellectual Property
(IP) rights (or there is potential IP)
e) Harm to participants (physical or psychological)
could be reported / discovered.
f) National policy might be influenced by your
findings or the results are historically important.
g) Any other reason that you may need access to
the data and/or samples in future.
